人类遗传分析中的新技术
当前位置:首页 >> 仪器仪表资讯
人类遗传分析中的新技术
人类遗传学的发展得益于过去十年中脱氧核糖核酸(DNA)分析技术的巨大进步。由于数据采集速率的大大加快,人们针对庞大的数据量发展了半自动化的方法用于原始数据采集、基因型分析和DNA直接测序。本文将介绍几种与实验设计和分析相关的最新进展,这些方法在家族遗传性致病基因的发现方面有着重要的应用前景。
1 方法概述
1.1 微卫星基因分型
微卫星标记,又叫短串联重复序列(STRs),由连续的单一序列重复单元(如CACACA...或者GATAGATAGATA...)构成[1]。人类基因组包含成千上万的该种核苷酸二倍、三倍和四倍重复序列[2]。围绕单一序列设计的聚合酶链反应(PCR)引物可以准确地扩增特异性微卫星序列。许多这样的位点都具有多态性,用于分隔具有众多重复长度的等位基因。这些PCR扩增产物可以用作对应染色体片段的遗传标记,使之能够按照家族进行分类。微卫星的长度通常在几个减数分裂期内保持不变,从而可以作为某些家族性疾病连锁分析的有效工具[3]。
目前微卫星的使用还面临着一些技术难题[4]。反复使用的PCR酶易于使合成的产物不保真,导致产生一系列比全长略小的序列,通常这些序列与原序列仅相差一个重复单位[5]。除此之外,更为严重的是这些酶可能在产物3′端添加一个额外的非模板核苷酸。如果额外增加的核苷酸发生了突变就会导致峰分裂现象的出现,从而产生没有实际应用价值的双核苷酸标记物(dinucleotide markers)。减少变异的一种方法是在一条PCR扩增引物5′端添加一个独特的序列标签。进行荧光基因分析时,这些带有标签的引物会作为非标记引物来使用(经常但不总是作为反向引物)。尽管这种序列标签的具体反应机理未明,但仍有几个标签已见报道[6,7]。
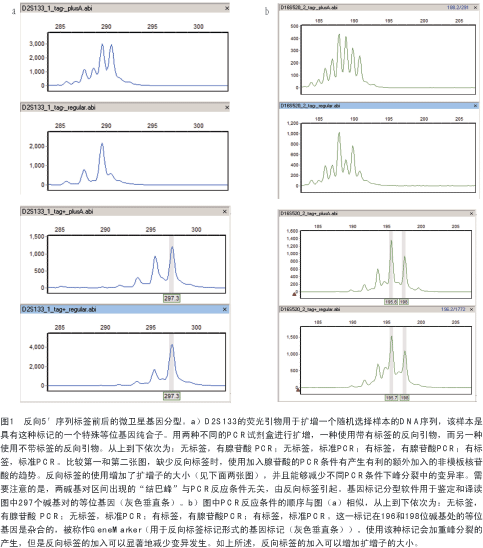
作者已经用特异添加腺苷酸的PCR和普通的标准PCR实验证实了这种序列标签的有效性。如图1a和b所示,两种不同双核苷酸标记物的差异在于:当使用默认设计的引物时,其对所用PCR试剂盒的敏感性不同。可是,如果在未标记引物中加入5′标签则可以减少两种标记物的变异,从而减少每一种标记物出现问题的几率。这提示我们:作为一种常规的防范手段,应当在所有常规的微卫星标记中加入这种标签。
1.2 SNP 基因分型
单核苷酸多态性(SNPs)和微卫星一样可用于遗传图谱的绘制。由于基因经常连锁在一起遗传,而连锁的基因并非完全随机地组成单体型,因此SNPs的内在信息量比微卫星携带的信息量要少些。然而协同分析的众多SNPs所携带的信息则类似或多于微卫星。作为人类基因组单体型图 (HapMap)计划的一部分,对几百万个SNPs进行了基因分型,并提供了几种不同人种的等位基因频率[8]。目前,用于SNP的高密度高通量芯片已经产业化,可用于传统家族性单基因紊乱的基因缺陷的遗传图谱绘制。
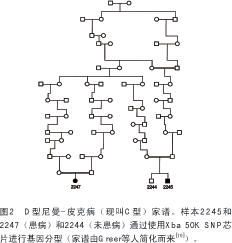
为了比较高通量SNP芯片和微卫星标记对家族性遗传图谱的有效性,作者利用大量不带D型隐性尼曼-皮克病的新斯科舍阿卡迪亚家族成员验证一个已知的遗传连锁[9,10]。通过原位克隆鉴定得知潜在的致病基因是NPC1(OMIM#257220)[11],从而在新斯科舍家族患病个体中证实了NCPI存在着纯合子突变[12]。作者又利用Xba 50K芯片(Affymetrix公司,圣克拉拉,加拿大)对两位患病的远房亲属和一位该家族未患病亲属进行基因分型(见图2)。如表1所示,两位患者 18号染色体上相同的等位基因中,具有共享纯合性的SNPs的最大拉伸长度约为7.7M碱基对,其中包括围绕NPC1的位于19.4M碱基对处的71个连续的SNPs。未患病亲属样本2244中有30个标记与患病者不一致,某些是杂合子而另一些是其他SNP等位基因的纯合子(该数据未显示)。由此可见 SNP芯片可以省时、省力、省钱地成功复制整个家族样品的连锁图谱。
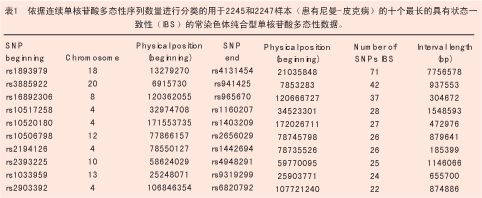
由于跨复合染色体的连续SNPs在绝对长度和数量上存在微小差异,所以作者用Xba和Hind芯片测定全部116 000个标记位点的分布。间隙长度的分配呈双模态分布,约在400和22 000碱基对处出现两个峰。除着丝粒外,2~3M碱基对长度之间存在3个间隙, 而1~2M碱基对长度之间还存在33个间隙。有些间隙处于末端着丝粒处,有些则位于基因相对稀少的区域,而其他的间隙可能由于信息SNPs附近缺少合适的抑制位点而处于基因密集区。 1~3M碱基对范围内的间隙数量提示我们应当充分注意对高密度连锁不平衡实验结果的解释。目前,最新出现的超高通量500K SNP板正变得越来越有用,对于某些实验设计而言,与一般的芯片相比其具有更大的优越性。
1.3 突变检测
基因发现实验的最终步骤是突变检测。尽管像变性高效液相色谱(dHPLC)等间接物理化学检测方法可以用于突变检测,但是DNA测序仍然是序列变异(也就是突变)检测的最权威的标准。手工检查大量序列信息不仅效率低下,而且由于人为误差容易遗失突变信息。几种半自动化方法已经被发展用于满足分子遗传分析逐渐增长的需要 [13-17]。
作者对用于序列变异检测的突变检测软件(MutSurv,SoftGenetics公司,州立学院,巴拿马)进行了评估。利用几个含有已知单碱基改变、插入和缺失变异的样本验证了该软件的有效性。该软件对样品标记进行排列和比较以提供参考或得到内在的一致序列标记和执行检测运算法则,并通过质量得分、不同色谱图和绘图输出等方式报告潜在突变/多态性。如果向其提供包括外显子和内含子位点在内的开放阅读框和已知变异,则可利用该软件输出和得到染色体组信息。
利用MutSurv软件对被测样品中所有已被鉴定的变异进行测定的结果在图3a和b中给出。在单核苷酸变异的情况下,软件能标出突变并方便地阐述该突变对潜在基因编码蛋白可能的影响。MutSurv软件在几种情况下都能鉴定出dbSNP数据库中标注的已知SNPs。通过对软件中沿外显子绘制的平面图与UCSC(加利福尼亚大学,Santa Cruz公司)基因组浏览器界面进行比较,可以非常容易地鉴定出已知的SNPs。在有插入和缺失的情况下,该软件不仅能够自动检测几个不同的突变,而且能对结果进行去卷积,从而准确地描述出插入和缺失的确切序列(见图3c),得到的结果可通过手工检查进行最终验证。

MutSurv软件标出了有问题的序列特别是阅读框末端附近这些序列中的几个假阳性结果。软件的新版本允许对末端进行剪切,从而减少假阳性的发生。尽管某些扩增子在正反两相上获得的序列可能存在问题,不过正反两种方向进行的突变检测仍然可以缩小低质量序列中假阳性的影响范围。
1.4 新的遗传计划
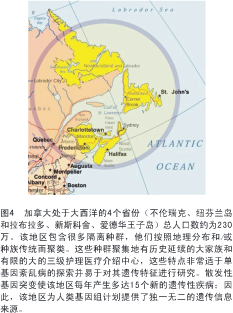
作者最近致力于从事总体范围内的基因探索,努力查明并从分子水平描述加拿大东部省份许多单基因人类紊乱病的特征(见图4)。这些工作采用本文描述的技术极大地提高了工作效率。将来可能也需要对这些技术进行一些改进,包括制备更高通量的SNP芯片;降低SNP芯片和 DNA测序的成本;以及减少个别先证者(指不依赖于家庭中其他成员而被独立检出的病例)整个基因组或整个外显子测序所需的最终成本。
2 方法应用
2.1 样品采集
利用标准方法从全血或者唾液中提取DNA。历史上除了抽血取样外还有口腔取样和采用血液污迹的方法,不过后两种方法很不稳定而且 DNA产量低下。利用The Oragene saliva kit (DNA Genotek公司,渥太华,安大略湖,加拿大)对样品进行测试。收集6个不同个体的样品,基因组DNA的产量为每人20~320μg不等(2 mL唾液样品)。通过凝胶电泳可知未消化的多是高分子质量的DNA。可能由于糖或脂质成分的残留导致某些样品存在轻度浑浊,A260/A280值为1.4~1.6(经A320校对)。通过对该方法进行微小改进,包括用70%酒精冲洗,就可以提高 A260/A280值。这样只需10 ng样品进行PCR反应就可以很好地用于微卫星基因分型和DNA测序。尽管作者没有对全基因组扩增产生的DNA长期储存和使用进行系统地评估,但应注意,已有厂家用Qiagen(巴伦西亚,加拿大)试剂盒对DNA进行重新纯化后用Affymetrix高密度SNP芯片证明了该方法的有效性。
所使用的全部样品均未违反相关的伦理制度,并征得了患者本人的同意。
2.2 微卫星基因分型
微卫星标记所需的引物可从GDB数据库获得。常规荧光微卫星标记由原始基因组序列使用Tandem Repeat Finder [18](与UCSC基因组浏览器结合[19,20])、Repeat Masker[21,22]和Primer 3[23]软件发展而来。每个得到的扩增子都带有一个标记荧光的正向引物和成对的未标记反向引物,该反向引物5′末端带或不带5′-GTTTCTT-3′ 序列标签都可以。对两种不同的PCR循环条件进行实验。普通的标准条件为:95 ℃(3 min)(循环1次);95 ℃(1 min),55~60 ℃(1 min),72℃(1 min)(循环30次);72 ℃(2 min)(循环1次)。特异添加腺苷酸的PCR反应条件为:95 ℃(5 min)(循环1次);94 ℃(15 s),55℃(15 s),72 ℃(30 s)(循环10次);89 ℃(15 s),55 ℃(15 s),72 ℃(30 s)(循环20次);72 ℃(30 min)(循环1次)[7]。扩增后用ABI 377在6%聚丙烯酰胺上进行电泳分析;再用ABI GenScan软件得到相应谱图(ABI 377和Gen-Scan购自应用生物系统公司,福斯特市,加拿大)。然后通过GeneMarker软件(SoftGenetics公司)对基因型谱图进行分析。基因型名称以文本形式输出并用PedCheck软件[24]进行遗传验证。
2.3 SNP基因分型
加拿大多伦多大学儿童医院的微阵列研究室通过Xba 50K SNP芯片对基因型进行收集。利用Affymetrix软件得到基因型的名称并以电子表格的形式从研究室输出。有58 960 个SNPs被收集,其中58 494个在人基因组中的位点是唯一的。纯合子SNP等位基因的长度可以通过对不同国家的数据库进行直接查询而得到,并对其物理位置或连锁纯合的状态一致性 (identical by state, IBS) 标记数量进行储存。结果中每个染色体着丝粒间隔的缺失序列和标记信息已被人工删除。
转速计| 水份计| 分析仪| 溶氧计| 电导度计| PH计| 酸碱计| 糖度计| 盐度计| 酸碱度计| 电导计| 水分测定仪| 浊度计| 色度计| 粘度计| 滴定仪| 密度计| 热流计| 浓度计|
2.4 突变检测
荧光DNA测序的扫描文件通过在ABI 377上进行电泳而获得。将该文件输入到Mutation Suveyor软件中以对序列变异进行分析。厂家的数据库地址或者国立生物技术信息中心(NCBI)可以提供基因组外显子/内含子和蛋白质编码信息。合成的野生型参照序列(wild-type reference sequence)扫描文件也可通过该软件由相同的基因组序列产生。
,更多相关信息
分类点击排行
- 1500米防水温度记录仪MX2204 HOBO TidbiT MX Tempe 5000
- HOBO无线蓝牙水下温度记录仪MX2203 TidbiT MX
- MX2201新款带蓝牙水下温度记录仪Pendant MX Temp
- 防火报警智能热像Fotric816/Fotric826
- 工程师通用热像仪Fotric 222s/Fotric 222s-1/Fotric 222s-2
- HOBO MX100蓝牙温度记录仪防水等级IP67
- 可程式噪音计TES-1352S带记录可插SD卡
- 数据记录型照度光量子计TES-1339P
- 便携一体式温湿度大气压力计TES-1160/TES-1161
- 高精度水位计MX2001-04-s带无线数据传输